今天阅读的文献是 2025 年 5 月安徽医科大学沈玉先教授研究团队发表在 Cell Death & Differentiation 上的一篇文献,名为 《Hepatic factor MANF drives hepatocytes reprogramming by detaining cytosolic CK19 in intrahepatic cholangiocarcinoma》。
肝内胆管细胞癌最初被认为起源于胆管上皮细胞的恶性转化,最近的研究显示肝细胞可以转化为成熟的胆管上皮细胞或形成功能性胆管的ICC细胞,但是其分子机制仍亟待研究。
中脑星形胶质细胞源性神经营养因子(MANF)是一种内质网(ER)应激诱导蛋白,最初被鉴定为神经营养因子新家族的成员,并在帕金森病、阿尔茨海默病、脑缺血和糖尿病中发挥细胞保护作用。研究表明,MANF可保护缺血或药物诱导的肝损伤,并通过负调控TLR4/NF-κB信号通路抑制外周炎症。MANF在肝细胞癌(HCC)中表达下调并抑制HCC进展,而在另一种原发性肝癌——ICC中则表达上调。然而,MANF 在 ICC中是否发挥促癌作用,以及MANF在成熟肝细胞转分化中的行为尚不清楚。
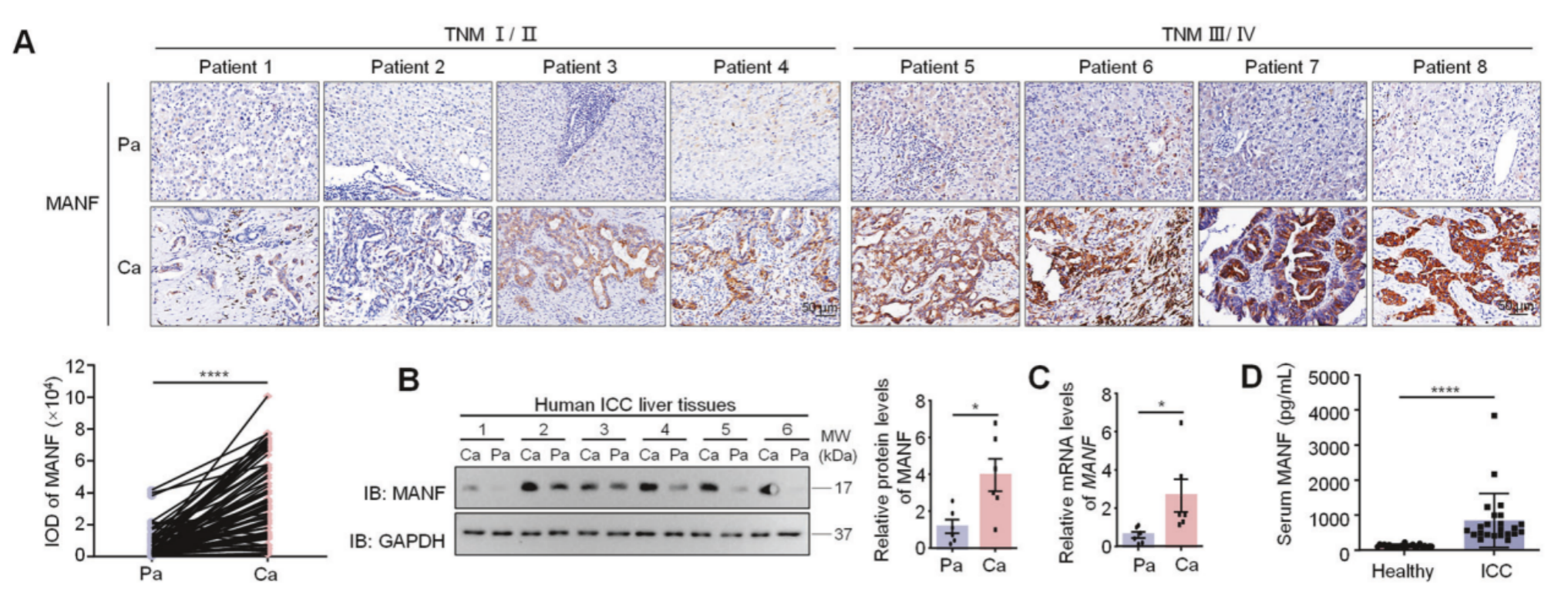
样本来源: 作者收集了78例单纯性ICC患者的石蜡切片、6例患者的冷冻样本(其中4例同时含有ICC和HCC组织),以及23例ICC患者和25例健康个体的血清。
分别使用免疫组化,WB,qPCR检测了癌与癌旁的MANF的表达水平,结果均显示ICC表达水平高。ELISA检测了健康者与ICC患者的血清中MANF的表达水平,ICC患者的表达水平高于健康者。作者也在公共数据库中进行了验证,生存曲线分析MANF的表达水平高预示着愈后不良,这些临床特征表明MANF是ICC诊断的预测性生物标志物。
随后作者构建了由HA-AKT、Myc-NICD1和转座酶组成的SBT系统小鼠ICC模型,结果显示注射SBT后小鼠肝脏肝脏体积增大,肿瘤数量和面积增多,HE染色显示ICC组织呈明显囊性腺样改变,SBT诱导后MANF在癌组织(Ca)中的表达显著高于癌旁组织(Pa),与载体对照组相比,SBT诱导的ICC组织中MANF的mRNA和蛋白水平均显著升高。
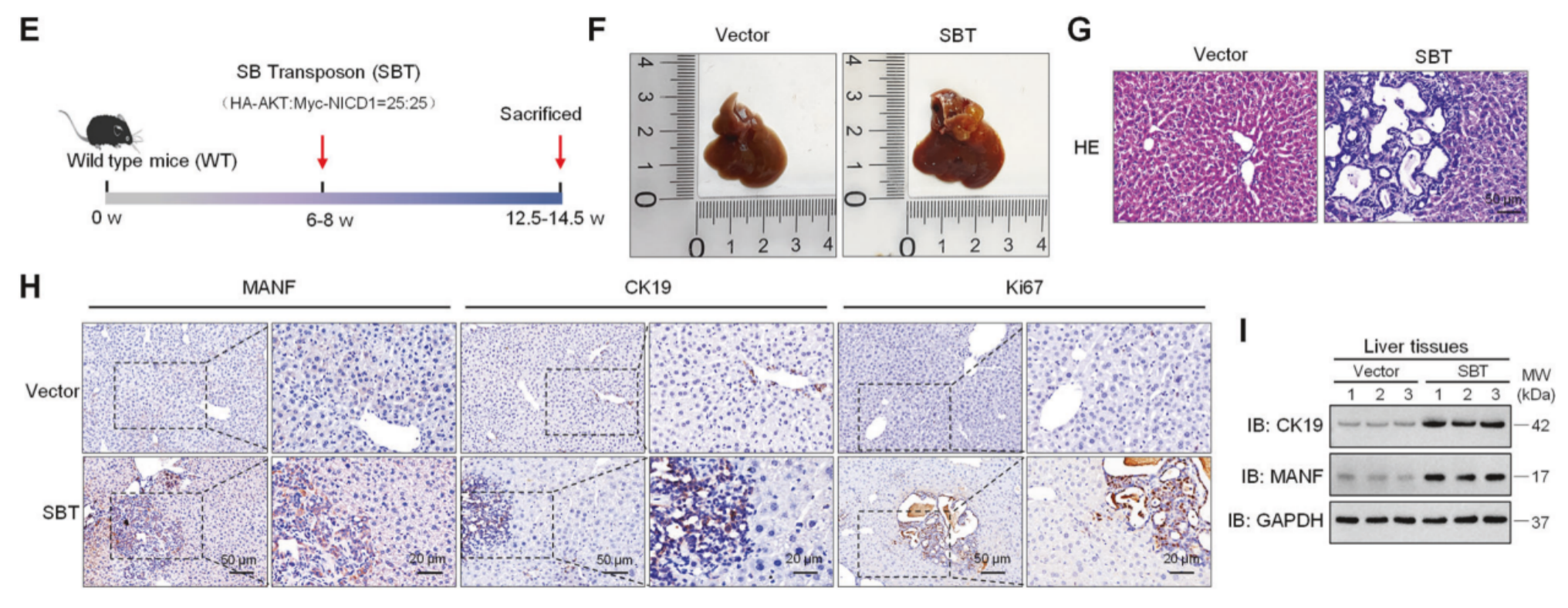
1该模型的原理和构建方法:
2睡美人转座子系统:(转座子:一个可移动的DNA片段,两端有特定的反向重复序列。)在本模型中,转座子上携带了两个关键的致癌基因:myr-AKT:表达持续激活形式的AKT激酶,能强力驱动细胞增殖和生存信号。NICD1 (Notch1 intracellular domain):表达Notch信号的持续激活形式,是驱动胆管细胞命运和癌变的关键因素。(转座酶:一种能识别转座子末端序列并将其“剪下”并“插入”到宿主基因组中的酶。)文献中常使用的有SB100或SB13,其中SB100的成瘤率更高(可达100%)
3
4构建方法:水动力尾静脉注射。
5制备质粒溶液:将携带致癌基因的转座子质粒和编码转座酶的质粒混合,溶解在大量生理盐水中(通常相当于小鼠体重的8-10%)。
6
7快速注射:在5-7秒内,将这个大体积的质粒溶液通过小鼠尾静脉快速注射进去。
8肝脏靶向:巨大的液体体积和注射速度会造成肝脏血窦的物理性扩张和通透性增加,使得质粒DNA得以高效地进入肝脏细胞。
9(通常在注射后4周左右就能形成肿瘤。其诱导的肿瘤在病理和分子特征上都与人ICC高度相似,例如表达胆管细胞标志物细胞角蛋白19(CK19),呈现腺样结构等)
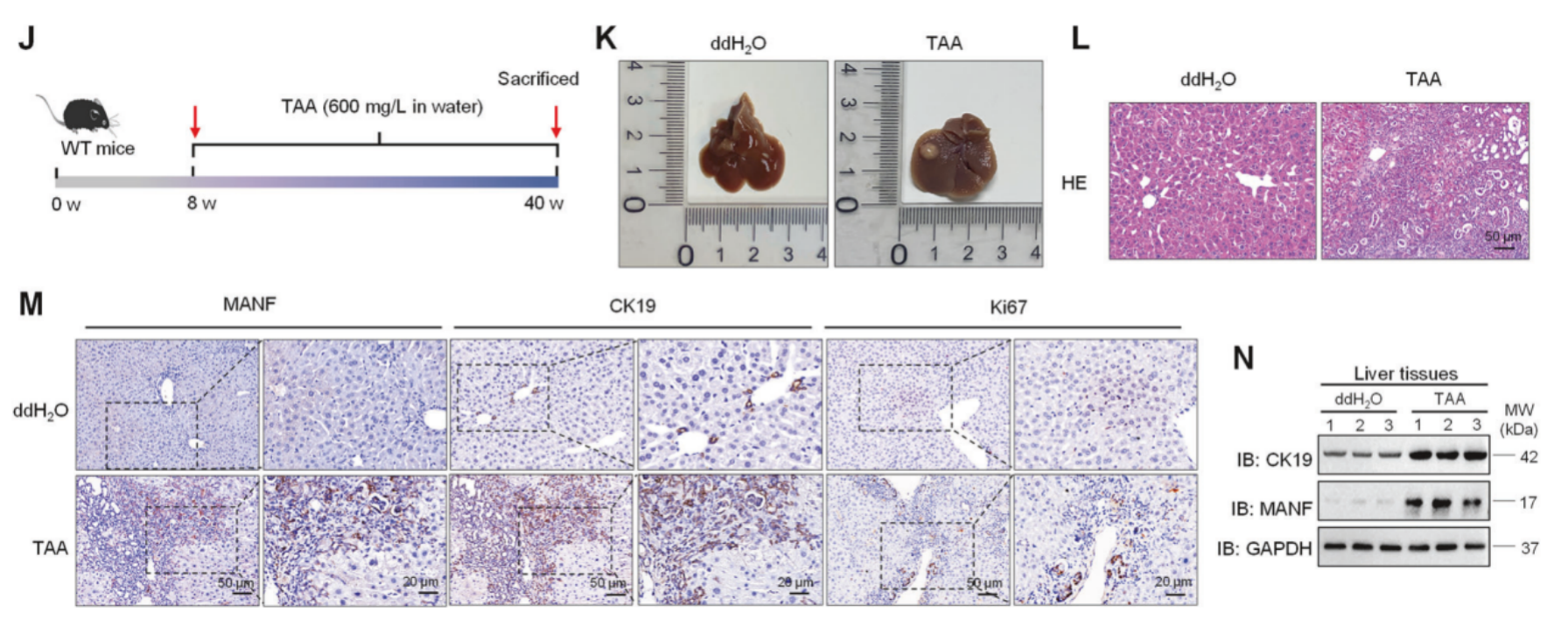
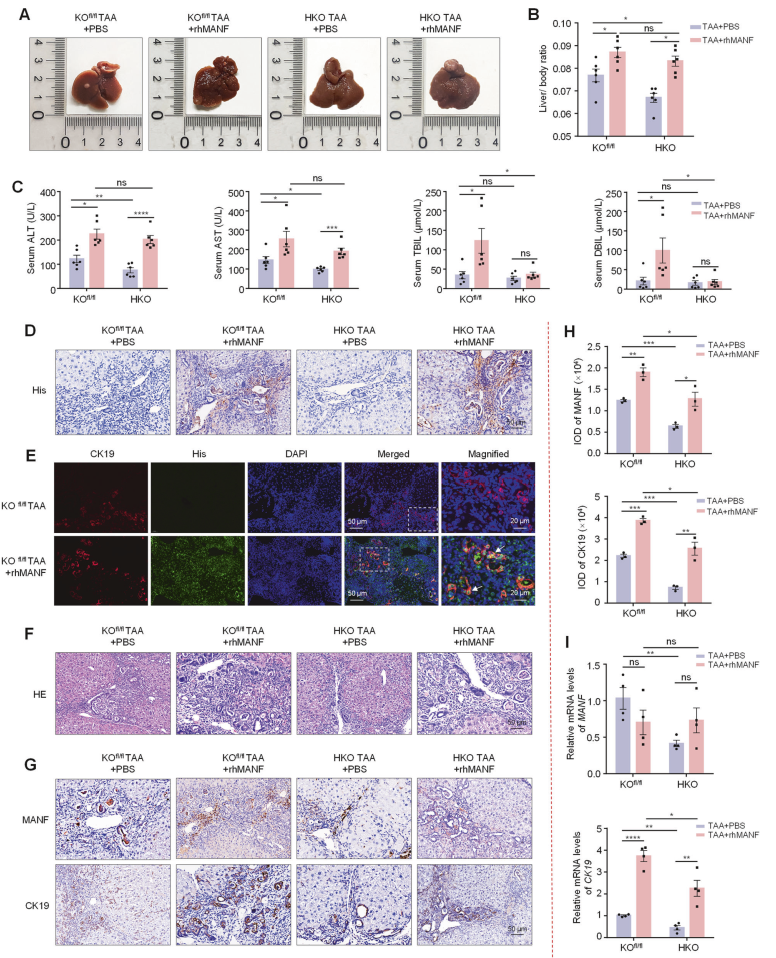
作者在TAA诱导的ICC模型中再次进行验证,TAA处理小鼠体重下降、肝体重比增加、血清ALT、AST、TBIL和DBIL水平升高、肿瘤结节和数量增多以及显著的胆小管反应和肿瘤面积增大,证实ICC模型诱导成功。在TAA诱导的小鼠ICC中,MANF在癌组织中的表达也显著高于癌旁组织。此外,TAA处理小鼠中MANF、CK19和Ki67的水平均升高,这与SBT诱导的小鼠ICC模型中的结果相似。
体外验证: 作者在Hucct1和RBE两种细胞系中敲低和过表达MANF,平板克隆,Transwell以及WB检测EMT指标,结果显示过表达MANF后,细胞的增殖能力增加,侵袭能力增加以及EMT转化增加,敲低则相反。
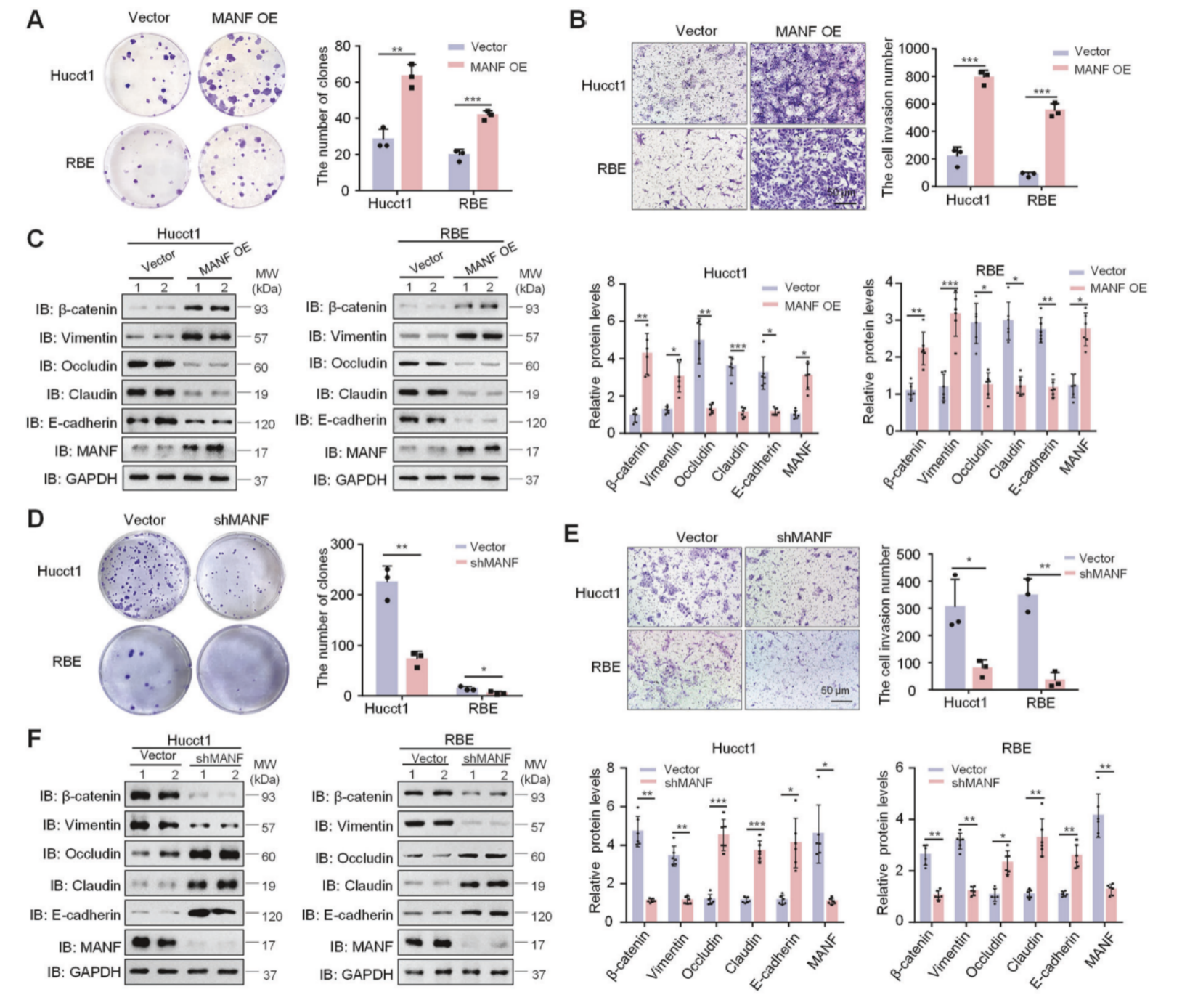
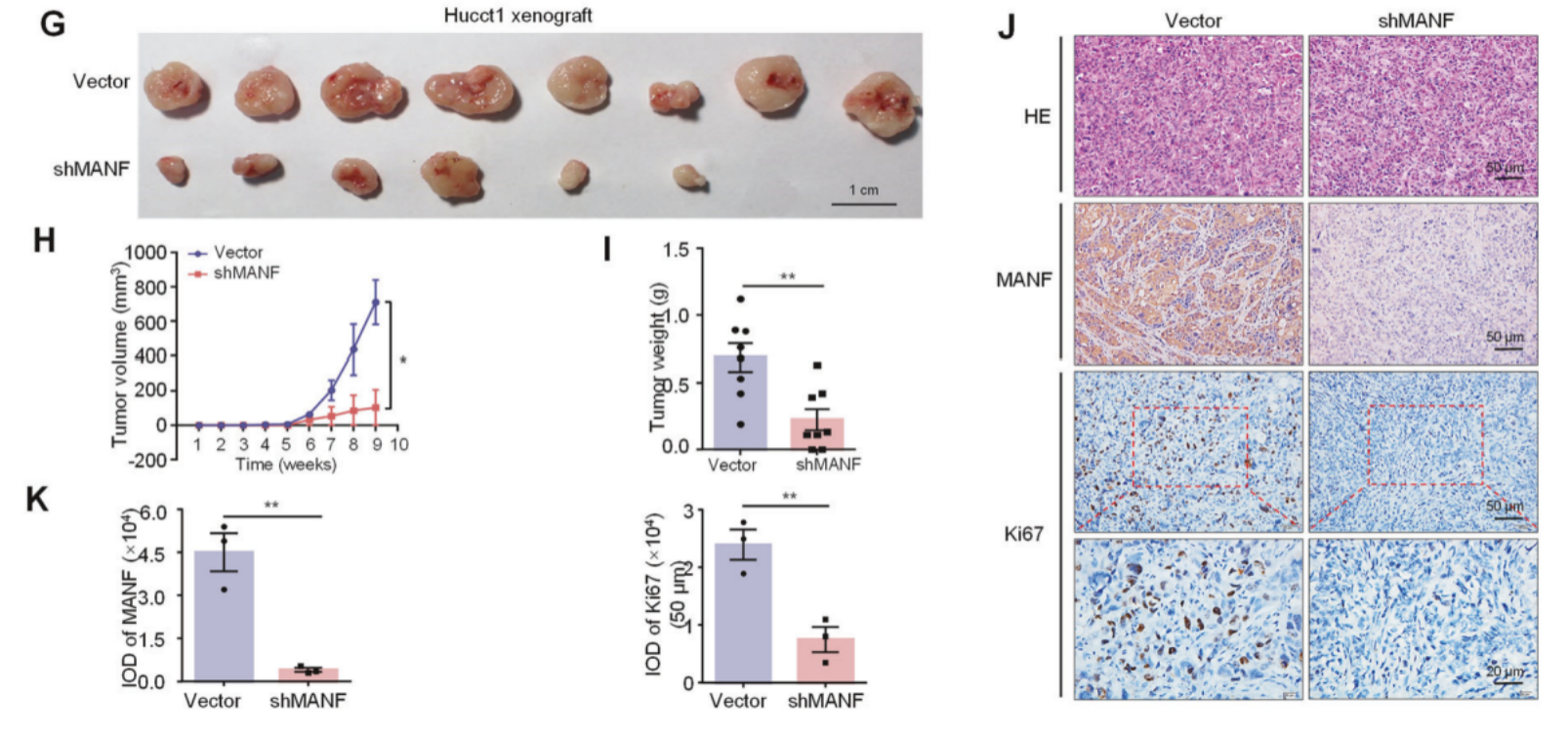
将上述的敲低细胞系进行小鼠的皮下成瘤,结果显示敲低MANF后小鼠的肿瘤减小,侵袭性减弱。
体内验证:小鼠模型1:SBT诱导的ICC
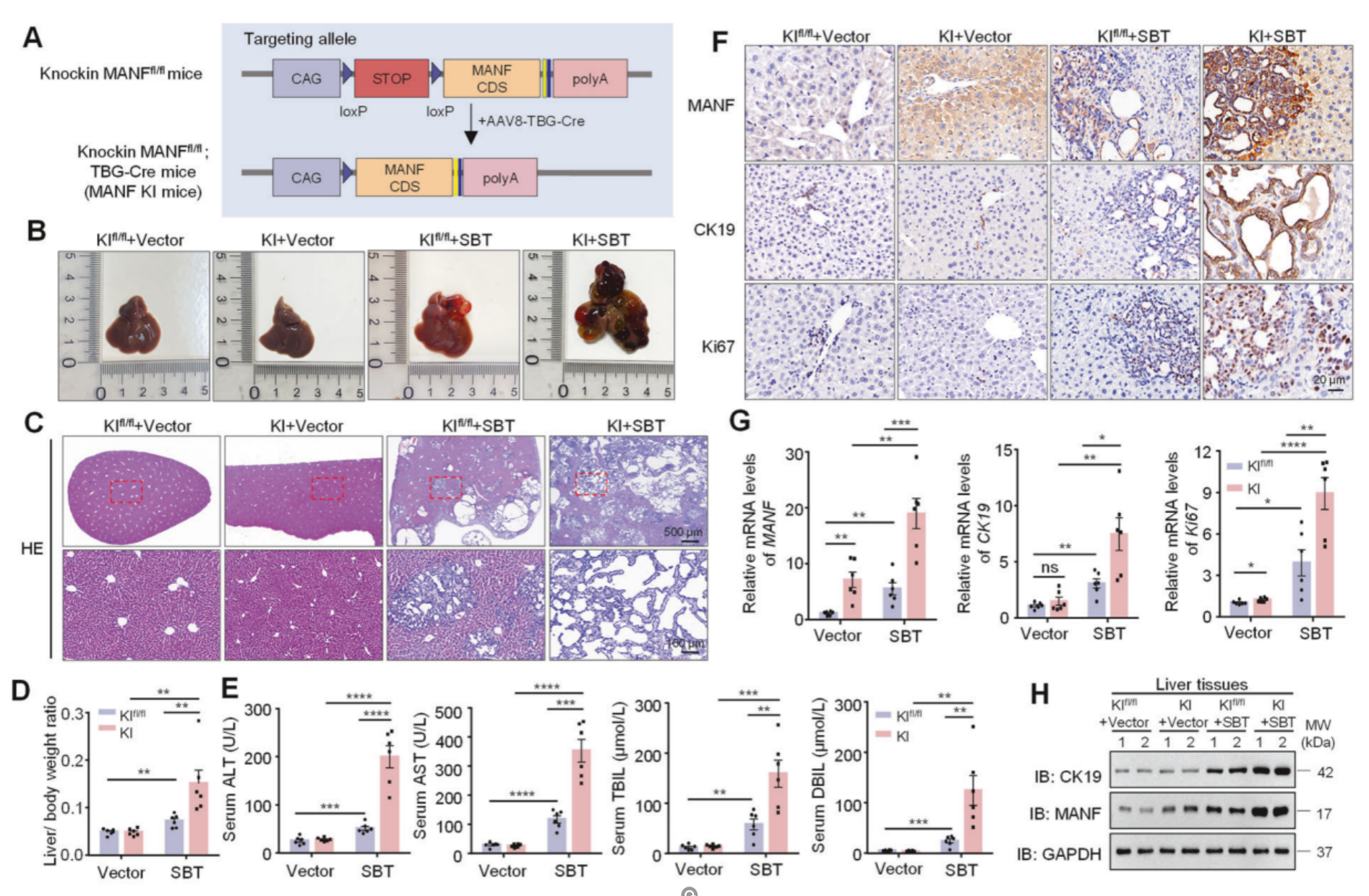
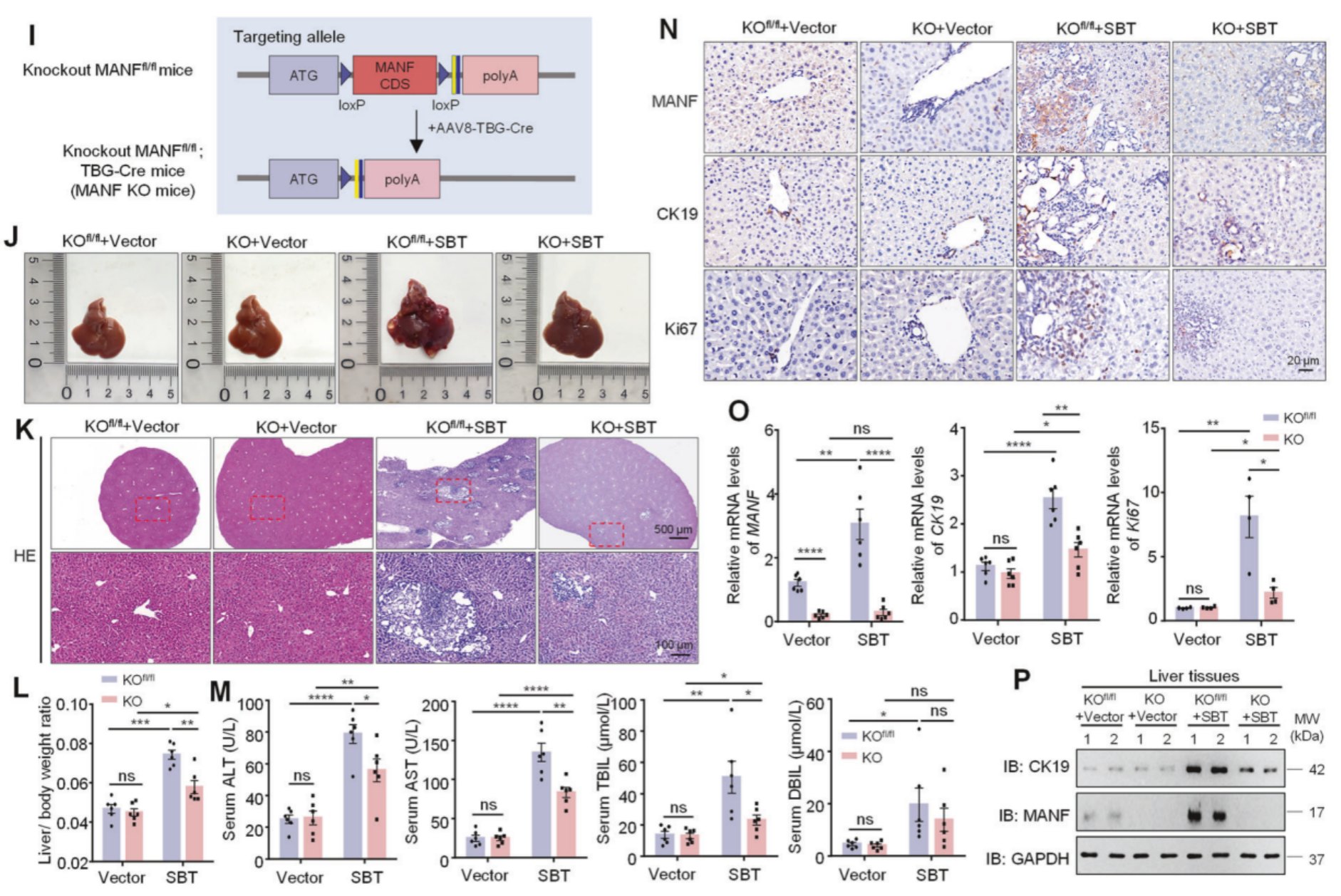
在通过CRISPR-Cas9技术,在AAV8-TBG-Cre调控下构建了成熟肝细胞特异性MANF敲入(KI)小鼠和MANF敲除(KO)小鼠,分别检测了小鼠的肝脏体积、肿瘤数量、肿瘤面积及肝体重;小鼠的血清ALT、AST、TBIL和DBIL水平;小鼠的MANF、CK19和Ki67的mRNA及蛋白水平。结果显示肝细胞特异性MANF敲入可促进小鼠ICC的发生与发展,敲除则抑制。
体内验证:小鼠模型2:利用Alb-Cre工具小鼠构建了肝脏MANF敲除(HKO)小鼠,再用TAA诱导ICC
用Alb-Cre工具小鼠构建了肝脏MANF敲除(HKO)小鼠。同时,对HKO小鼠给予rhMANF处理,以观察MANF缺失后补充MANF的挽救效应。重组MANF通过尾静脉注射给药,每周两次,剂量为2 mg/kg,持续28周。通过抗His抗体的免疫组化和免疫荧光检测rhMANF在肝组织中的分布。结果发现,在TAA诱导下,与KOfl/fl小鼠相比,HKO小鼠的肿瘤大小和数量、肝体重比、血清ALT和AST水平以及肿瘤面积均显著减小,而这一效应被rhMANF所抑制。我们还发现,在经TAA处理的KOfl/fl小鼠中,与PBS对照组相比,rhMANF处理增加了肝体重比、肿瘤数量和面积以及血清ALT和AST水平。rhMANF处理后,CK19的mRNA和蛋白水平均升高。这些数据进一步表明,肝脏MANF敲除可抑制ICC,而补充rhMANF可促进ICC并抵消MANF敲除对ICC的作用。
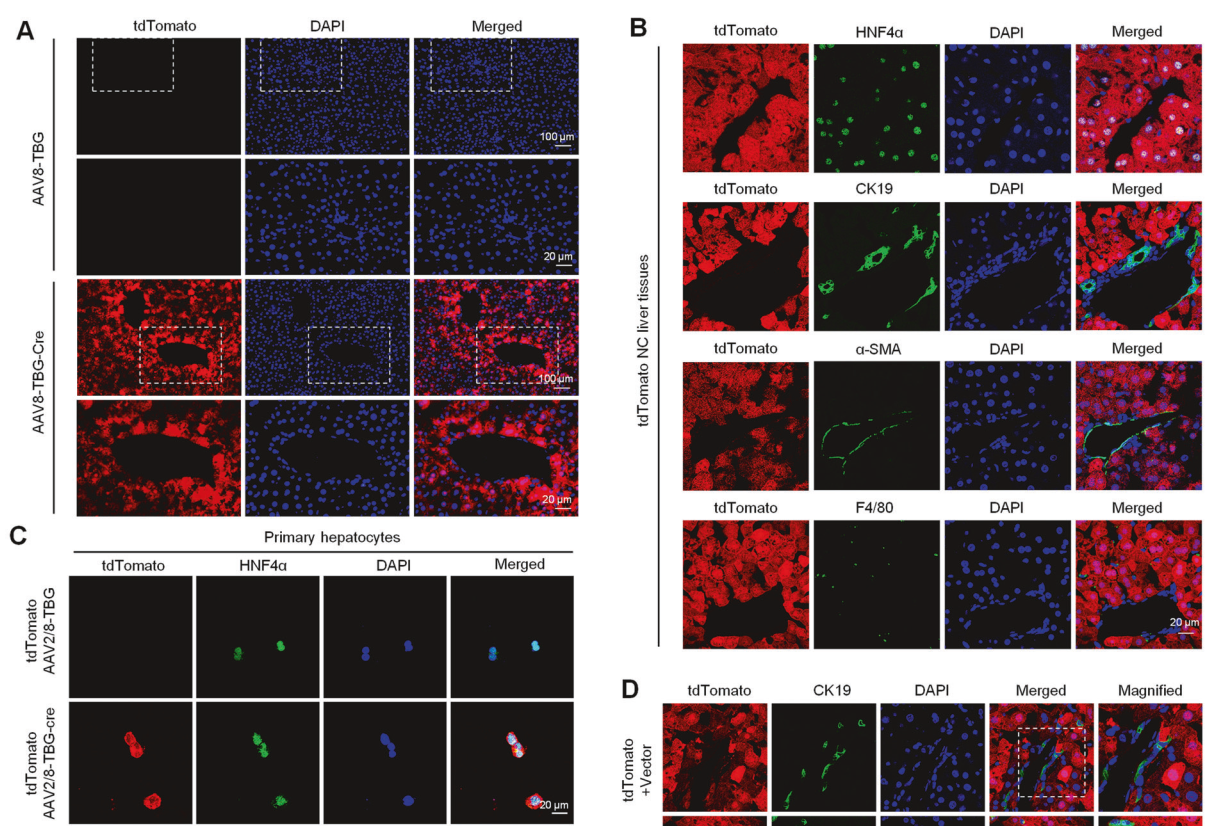
在小鼠层面看正常肝细胞发育谱系,以及在SBT和TAA诱导下的肝细胞发育谱系
通过注射AAV8-TBG-Cre的tdTomato荧光报告小鼠实现了成熟肝细胞的谱系示踪。结果显示tdTomato示踪的肝细胞。我们验证了tdTomato在肝组织中特异性地表达于HNF4α标记的肝细胞,而不表达于CK19标记的BECs、α-SMA标记的肌成纤维细胞和F4/80标记的巨噬细胞。
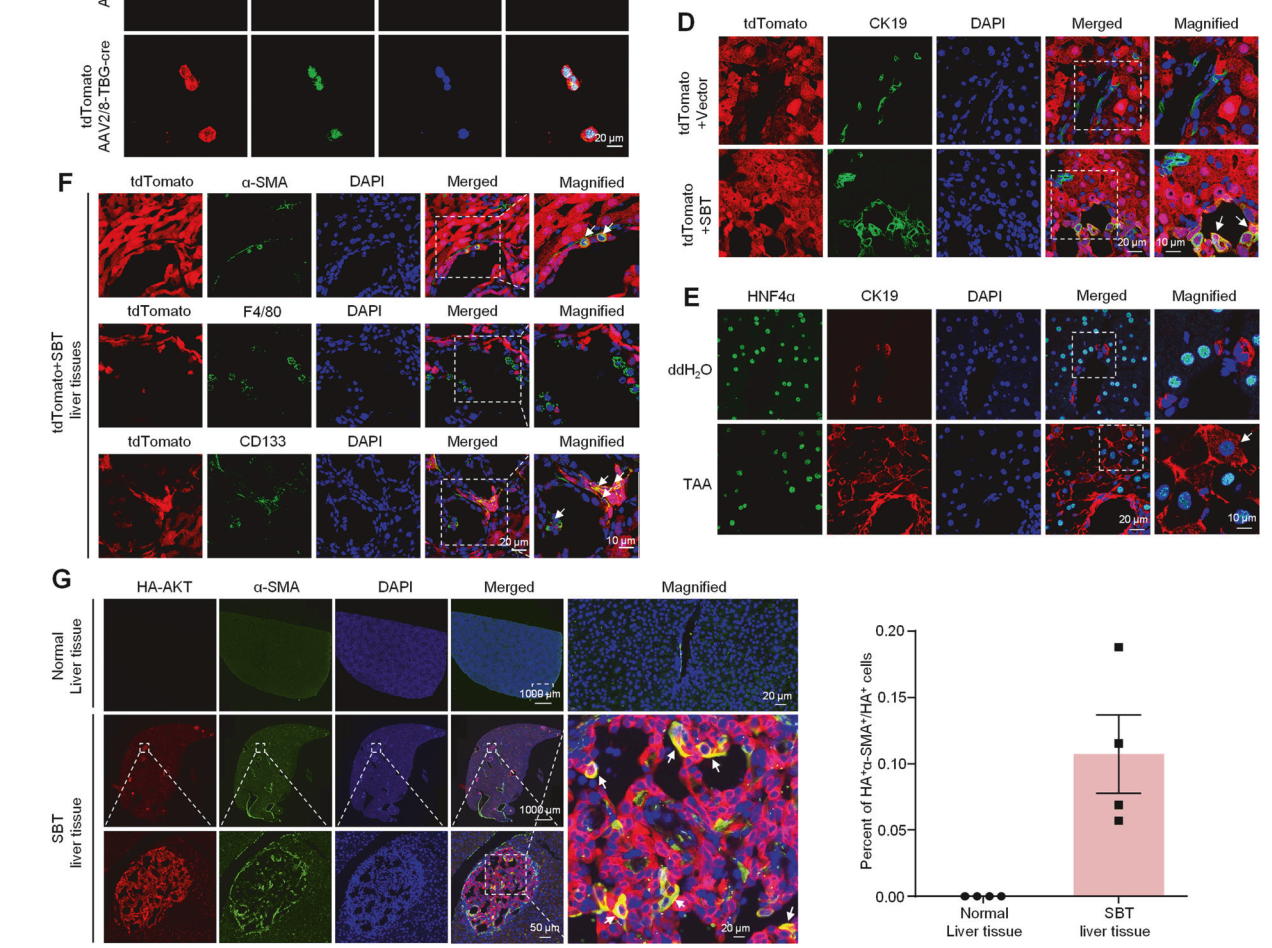
随后利用tdTomato小鼠制备SBT诱导的ICC。在SBT诱导的ICC小鼠中可检测到tdTomato与CK19的共定位,而载体对照组中则无此现象(图5D),表明SBT攻击后肝细胞被重编程为ICC细胞。在TAA诱导的ICC中也发现了HNF4α+CK19+细胞(图5E)。我们还发现成熟肝细胞转化为CD133+干细胞(肿瘤干细胞)和α-SMA+肌成纤维细胞,但未转化为F4/80+巨噬细胞(图5F)。进一步研究表明,SBT攻击后约有11%的HA+肝细胞转化为α-SMA+肌成纤维细胞(图5G)
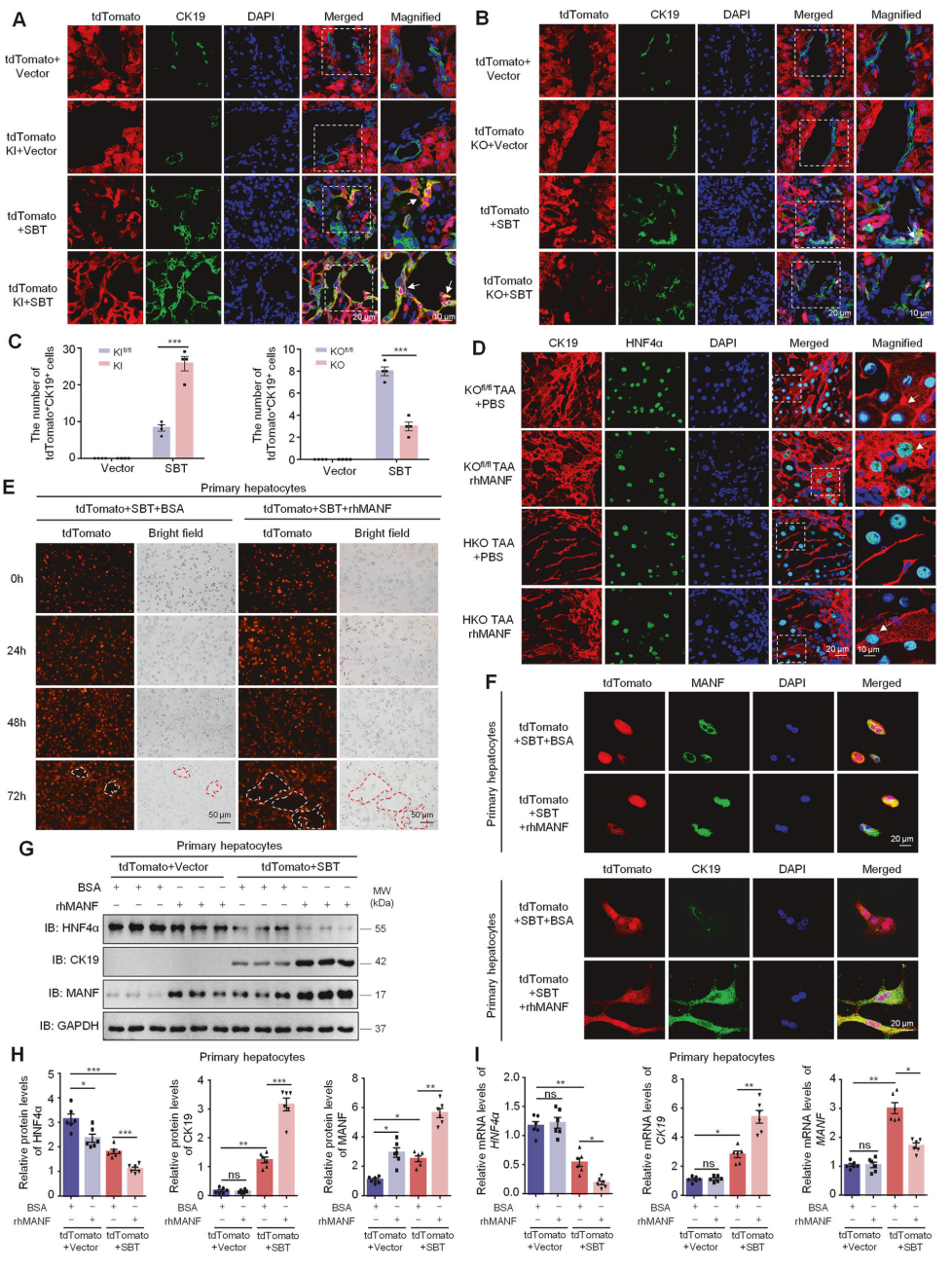
在基因小鼠层面看MANF对肝细胞发育的影响
通过SBT诱导ICC模型,SBT注射后,肝组织中出现了tdTomato+CK19+双阳性细胞,且MANF KI小鼠中的双标记细胞多于对照小鼠,相反,MANF KO小鼠中tdTomato+CK19+细胞较对照小鼠减少。
在TAA诱导的ICC小鼠中发现了许多HNF4α+CK19+细胞,且rhMANF处理后CK19水平升高。然而,在TAA诱导的ICC小鼠中,HKO小鼠的HNF4α+CK19+细胞数量减少,而这一效应在rhMANF处理后得到逆转。这些结果表明,MANF驱动成熟肝细胞向ICC细胞转化。
从经SBT攻击2周的tdTomato小鼠中分离原代肝细胞。以80 ng/mL浓度的rhMANF处理原代肝细胞。与BSA处理的对照组相比,rhMANF处理的tdTomato+肝细胞中观察到明显的导管样结构形成。此外,rhMANF补充不仅增加了细胞内MANF水平,还增加了tdTomato+CK19+细胞数量及胞质CK19水平。我们还发现,无论是否注射SBT,rhMANF处理均下调原代肝细胞中HNF4α的表达,而仅在SBT暴露后才上调CK19。原代肝细胞中HNF4α和CK19的mRNA水平变化与其蛋白水平变化一致。然而,MANF mRNA水平在SBT暴露后明显升高,但在rhMANF处理后降低。
这些数据表明,rhMANF作为外源性诱导剂,促进成熟肝细胞谱系重编程为ICC细胞。
在TAA诱导的ICC小鼠的HNF4α+CK19+双阳性细胞中,CK19主要定位于细胞膜。而经rhMANF处理后,双标记细胞中的CK19主要定位于细胞质。由此现象去看MANF对CK19定位的影响
随后作者在细胞系中过表达MANF,发现总的CK19增加,但是胞质中CK19增加更明显,MANF增加,胞质增加水平高于胞膜。
MANF和CK19的免疫荧光双标记也显示,在肝组织和原代肝细胞中,MANF与CK19的共定位主要发生在细胞质中。
MANF与CK19之间的相互作用通过Co-IP、GST-pull down和Biacore实验得到验证
找具体结合结构域并且做了该结构域的功能实验
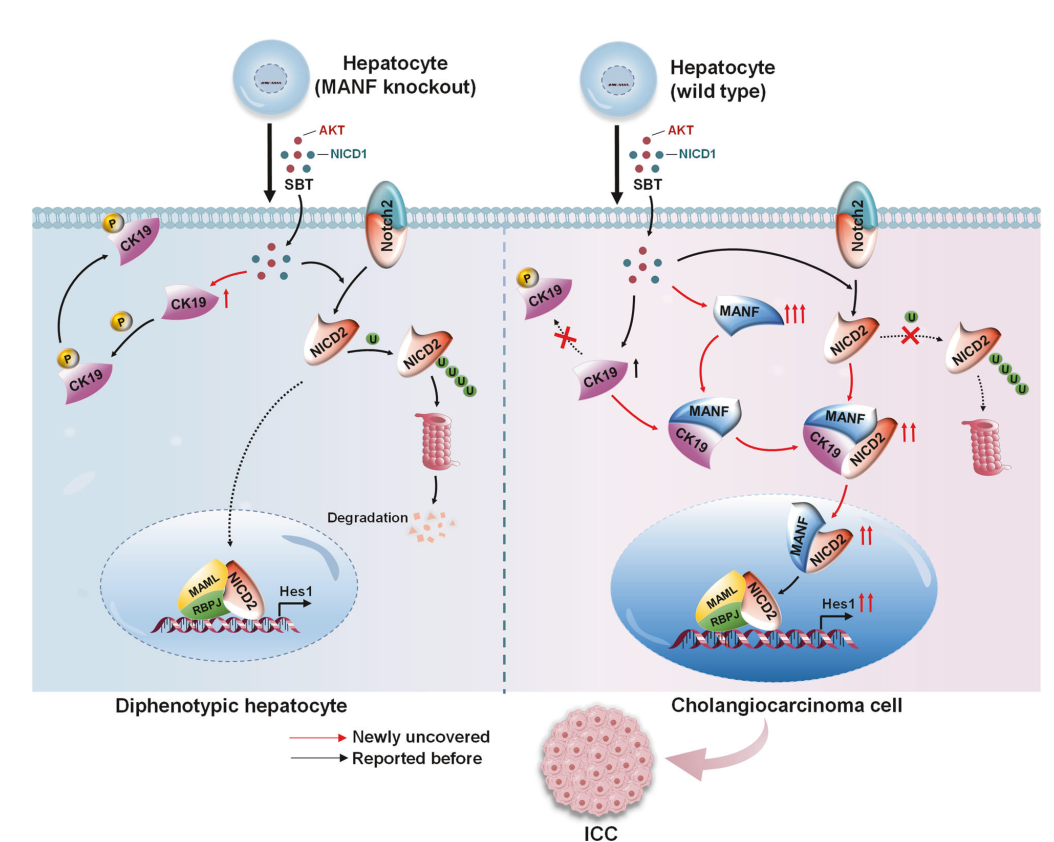
鉴于CK19在Ser35位的磷酸化对其膜募集至关重要,我们将CK19的Ser35突变为Ala35(CK19S35A),结果发现CK19突变取消了MANF与CK19之间的相互作用(图7G)。当我们在Hucct1细胞中过表达HA-CK19S35A时,与野生型相比,膜上CK19水平降低,而胞质中CK19水平升高(图7H、I)。这些结果表明,MANF在ICC细胞中通过与CK19的Ser35相互作用,抑制CK19的膜转位。
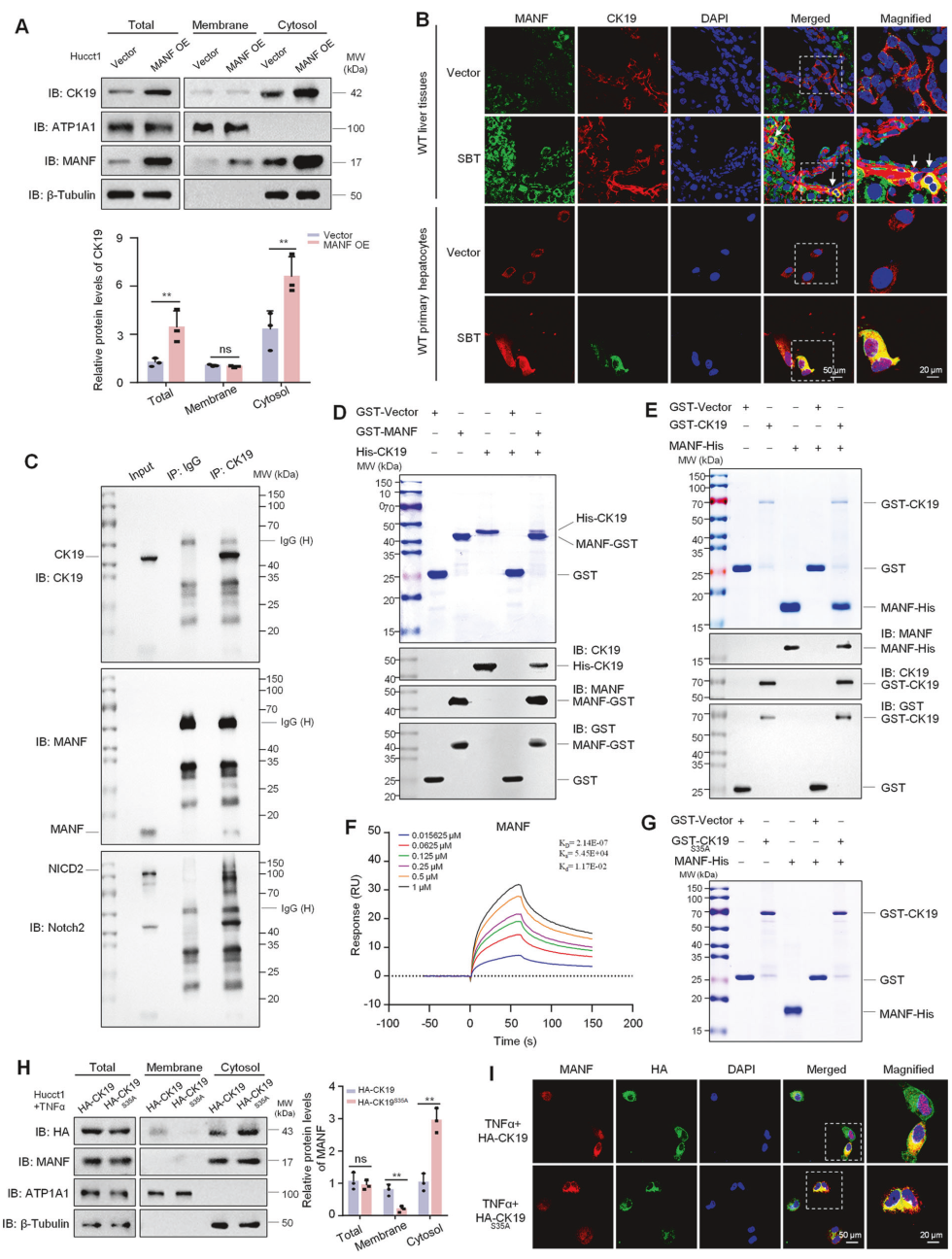
接下来就去探索CK19在ICC中发挥着什么作用,CO-IP联合质谱找到了CK19相互作用蛋白Notch2。。CK19与Notch2之间的相互作用通过Co-IP、GST-pull down和Biacore实验得到验证。此外还构建了Notch2的截短体,包括胞内结构域(NICD2)及其各组分。Pull down实验显示,CK19与NICD2及AR结构域相互作用,但不与RAM、TAD和PEST结构域相互作用。
在经SBT处理2周的ICC小鼠分离的原代肝细胞中,发现MANF敲除降低了胞质CK19水平。同时,MANF敲除降低了胞质和核中的NICD2水平。相反,MANF过表达增加了胞质CK19和NICD2以及核内NICD2水平。为明确CK19对NICD2稳定性的影响,在过表达HA-CK19的Hucct1细胞中进行了放线菌酮(CHX)追踪实验。结果发现,CK19过表达显著增加了Notch2的稳定性并延长了其半衰期.
从SBT诱导的ICC小鼠分离的原代肝细胞中Notch2及其下游分子的表达。随着CK19和Notch2的增加,MANF敲入后Hes1、Jag1和CK7水平升高,而MANF敲除后则降低.
为进一步确定MANF、CK19和Notch2在ICC中的关系,使用多重免疫荧光染色检测了ICC与HCC混合肝组织切片中MANF、CK19和Notch2的表达。MANF、CK19和Notch2的共定位仅在ICC中观察到,而在HCC中未观察到。GST-pull down实验揭示了MANF、CK19和NICD2的复合物(图8J),提示MANF可能作为连接CK19和NICD2的桥梁。
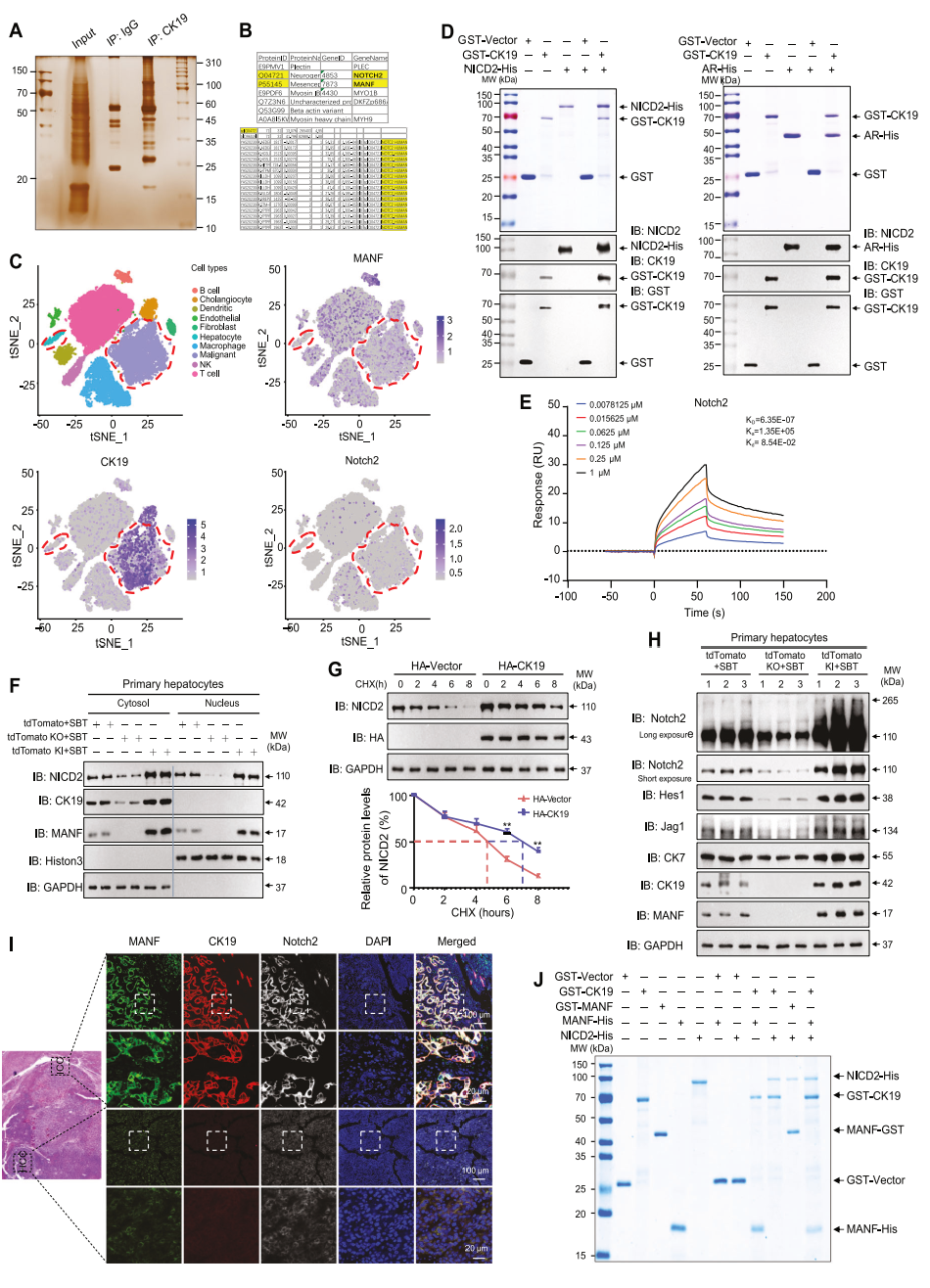
为探究NICD2是否单独在MANF调控肝细胞向ICC细胞转分化中发挥关键作用在小鼠肝中Notch2沉默,与对照组相比,经SBT处理的小鼠肝脏明显缩小,肿瘤数量、面积及肝重比也显著降低(图9B-F)。由MANF过表达诱导的CK19、Ki67、CK7上调及HNF4α下调可被Notch2敲低所逆转(图9G、H、J)。
谱系示踪结果显示,MANF KI引起的tdTomato+CK19+细胞增加可被Notch2沉默部分挽救(图9I),这充分表明MANF促进肝细胞向ICC细胞转化的作用部分依赖于Notch2。
同样,MANF过表达诱导的Notch2和CK7上调也可被CK19沉默部分抑制(图9K)。同时, Hucct1细胞中沉默MANF可降低CK19与NICD2之间的相互作用(图9L),提示MANF增强CK19-NICD2相互作用。进而,MANF增强NICD2稳定性及Notch2激活,从而促进肝细胞重编程为ICC细胞。这些结果表明,MANF促进肝细胞向ICC细胞重编程的作用部分依赖于CK19和Notch2。
在本研究发现MANF在ICC组织中高表达,且与ICC患者的生存相关。其次,证明了MANF在ICC中发挥促癌作用。此外,通过使用荧光报告小鼠发现了MANF在SBT和TAA诱导的ICC中促进成熟肝细胞向胆管细胞样细胞转化。其潜在机制包括:MANF物理结合CK19的Ser35位点,抑制CK19的膜转位;MANF促进与CK19和NICD2形成复合物;胞质CK19与NICD2的AR结构域相互作用,抑制NICD2降解并激活其核信号,从而加速成熟肝细胞向ICC细胞的转化。